Spezialgebiet Vaskulitiden
Vaskulitiden: Drei wichtige Erkrankungsgruppen im Überblick
Vaskulitiden umfassen verschiedene Entzündungen der Blutgefäße, die je nach betroffenem Gefäßtyp und Altersgruppe unterschiedliche Symptome verursachen. Die Riesenzellarteriitis betrifft vorwiegend Menschen ab 50 Jahren und kann sich als Schläfenarterien-Entzündung, Polymyalgia rheumatica oder Großgefäßvaskulitis manifestieren. Bei allen Formen drohen ohne schnelle Cortisontherapie schwerwiegende Komplikationen wie Erblindung.
Die ANCA-assoziierten Vaskulitiden befallen kleine Blutgefäße und können einzelne Organe wie Niere oder Lunge schädigen. Zu dieser Gruppe gehören die Granulomatose mit Polyangiitis, mikroskopische Polyangiitis und eosinophile Granulomatose mit Polyangiitis. Der Nachweis spezifischer Antikörper (ANCA) im Blut hilft bei der Diagnose, wobei neue Medikamente wie Avacopan die Behandlungsmöglichkeiten erweitern.
Das Behçet Syndrom ist ein "Chamäleon" unter den Vaskulitiden, das alle Gefäßtypen befallen kann und besonders junge Erwachsene gefährdet. Die Diagnose kann aufgrund atypischer Verläufe Jahre dauern, doch das charakteristische Pathergie-Phänomen und das gute Ansprechen auf TNF-Hemmer sind wichtige diagnostische und therapeutische Hinweise.

Die Riesenzellarteriitis (RZA) ist eine Entzündung der Gefässwand von mittelgrossen und grossen Arterien. Sie befällt ältere Menschen ab 50 Jahren, mit kontinuierlichem Anstieg der Erkrankungswahrscheinlichkeit bis ins hohe Alter. Es gibt drei Formen der Erkrankung:
- Entzündung der Schläfenarterie; sogenannte Arteriitis temporalis
- Polymyalgie rheumatica (PMR)
- Grossgefässvaskulitis
Die Arteriitis temporalis führt zu neuartigen Kopfschmerzen im Schäfenbereich oder im Nacken; Sehstörungen wie Doppelbilder, Erblindung; Schmerzen in der Kaumuskulatur beim Essen; Empfindlichkeit der Kopfhaut.
Die Polymyalgia Rheumatica äussert sich in Muskelschmerzen im Schultergürtel und im Beckengürtel. Betroffene fühlen sich krank und beim Aufwachen am Morgen steif, sie können die Arme kaum heben.
Die Grossgefässvaskulitis führt zu ausgeprägtem Krankheitsgefühl mit Gewicht- und Appetitverlust und Fieber. Die Beschwerden lassen an eine Infektion (Blutvergiftung) denken. Es handelt sich um eine Entzündung der Gefässwand der Hauptschlagader und der grossen Gefässe im Brust- und Bauchraum (Large Vessel Vasculitis LVV).
Abklärungsschritte beinhalten:
Blutuntersuchung, welche meist hohe Entzündungswerte zeigt Gewebeprobe (sogenannte Biopsie) einer Schäftenarterie (kleiner chirurgischer Eingriff) oder mittels einer bildgebenden Untersuchung:
- Ultraschalluntersuchung der arteriellen Gefässe am Kopf
- Kernspin-Untersuchung (MR-Angiographie)
- PET-CT (Positron-Emissions-Tomographie verbunden mit Computertomographie)
Therapie: Der sofortige Therapiebeginn mit Cortisonpräparaten (in der Regel Prednisonpräparat) ist entscheidend, um einer möglichen Erblindung bei Befall der Kopfgefässe vorzubeugen. Kontaktieren Sie im Notfall eine der Telefonnummern / Adressen der VASAS in Ihrer Region!!
Die ANCA-Assoziierten Vaskulitiden (AAV) betreffen die Wand der kleinen Blutgefässe. Im Gegensatz zu den Grossgefässentzündungen wie der RZA oder der TAK können bei den ANCA-Vaskulitiden einzelne Organe wie die Niere oder die Lunge betroffen sein. PatientInnen sind im Mittel um 50-jährig, allerdings können auch Kinder und Betagte daran erkranken.
Zu den AAV gehören die
- Granulomatose mit Polyangiitis [GPA; früher Morbus Wegener genannt],
- Mikroskopische Polyangiitis [MPA]
- eosinophile Granulomatose mit Polyangiitis [eGPA; früher Churg Strauss Syndrom genannt].
Der Begriff ANCA+ besagt, dass ein Antikörper im Blut gegen ein Eiweiss von weissen Blutkörperchen gefunden wird (ANCA = Anti-Neutrophiler Cytoplasmatischer Antikörper). Dieser Antikörper spielt in der Krankheitsentstehung eine Rolle.
GPA: Erste Symptome treten typischerweise im Nasen-Nasennebenhöhlen-Bereich auf. Die Entzündung führt zu meist blutiger Sekretion, sie wandert entlang den Luftwegen in die Lunge. Diese lokalisierte Form der GPA ist oft ANCA-negativ. Sind andere Organe wie die Niere oder das Nervensystem betroffen, so ist der ANCA praktisch immer nachweisbar und gilt es, eine Therapie sofort zu starten.
eGPA: Die seltenste Form der AAV entwickelt sich auf dem Boden einer allergischen Bronchialerkrankung. Wenn die glucocortikoidhaltigen Nasensprays nicht mehr ausreichen und mit Prednisontabletten behandelt werden muss, so kann dies Zeichen eines EGPA-Beginns sein.
MPA: Ist die hinterlistigste Form der AAV, denn es fehlen Warnsymptome wie Asthma oder chronische Nasen- Nasennebenhölen Entzündung. Hinterlistig auch, da oft die Niere betroffen ist und diese keine Symptome wie Schmerzen oder Entzündungszeichen verursacht.
Die Abklärung der AAV umfasst Blut- und Urinuntersuchungen und eine systematische Prüfung aller Organsysteme. Wenn möglich sollte eine Gewebeprobe entnommen werden, um die Diagnose zu sichern. Bei allen Formen ist ein rascher Behandlungsbeginn wichtig, um Organschäden zu verhindern.
Neue Medikamente:
Es sind neue, zum Teil neuartige Medikamente verfügbar, welche grosse Fortschritte in der Behandlung bedeuten (für Infos über Zulassung siehe www.compendium.ch):
Avacopan: (Tanvenos®): Neuartiger Therapieansatz: Blockiert einen Complementrezeptor (C5aR1) und greift damit in die Entzündungskaskade ein. Vor allem bei schwerem Nierenbefall verwendet. Spart Glucokortikoide (e.g. Prednison)
Mepolizumab: (Nucala®) und Benralizumab: (Fasenra®): Blockieren die Wirkung des wichtigsten Wachstumsfaktors von eosinophilen Granulozyten, die eine zentrale Rolle im Krankheitsmechanismus spielen. Hilft wesentlich, Cortisonpräparate zu sparen (e.g. Prednison)
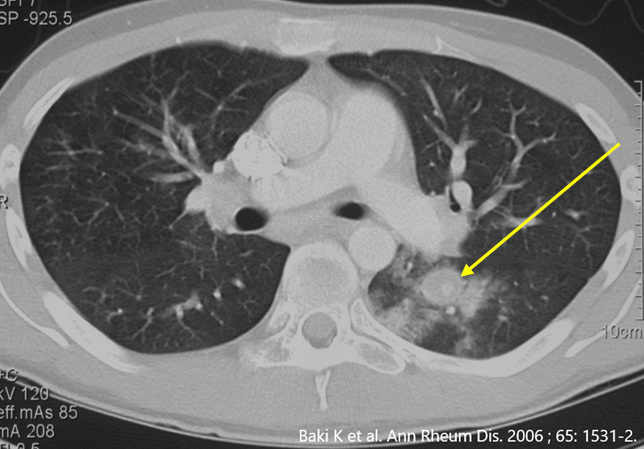
Das Behçet Syndrom (BS) ist ein Chamäleon. Bei klassischer klinischer Präsentation ist das BS einfach zu diagnostizieren, bei atypischer Krankheit kann es zu einer jahrelangen Verzögerung bis zur Diagnose kommen (siehe Publikationsliste). Das BS kann alle Gefässtypen befallen, von grossen Arterien über mittlere und kleine Gefässe bis zu grossen Venen wie die Hohlvenen des Rumpfes. Die gefährlichsten Formen finden sich im jungen Erwachsenenalte, beispielsweise eine Entzündung der Lungengefässen (*), der Beinarterien oder ein Hirnbefall.
Das BS hat eine Eigenart, die betreffend Therapie wichtig ist, nämlich das sogenannte Pathergie-Phänomen. Dieses beschreibt, dass bereits ein kleines Trauma wie ein Nadelstich eine örtliche Entzündung auslösen kann. Folgenschwer kann diese Eigenschaft bei Gefässoperationen sein, denn unbehandelt führen Gefässoperationen oft zu teils schweren Komplikationen und Re-Operationen.
Interessant ist die Tatsache, dass das BS fast ausnahmslos gut auf TNF-Hemmung (Infliximab, Adalimumab, etc) anspricht. Falls ein vermutetes Behçet Syndrom nicht auf TNF-Hemmer anspricht, so gilt es die Diagnose zu überprüfen. Das gute Ansprechen wird in der Abbildung über ein Hautgeschwür illustriert. Nach einer einzigen Injektion von Adalimumab (40mg) verschwand das chronische Geschwür und die geplante Operation konnte annulliert werden.
Informationen über die BS in der Schweiz finden sie in einer unserer Publikationen: (Behçet’s syndrome: clinical presentation and prevalence in Switzerland; Villiger Rahel A., et al. Swiss Med Wkly. 2019;149:w20072. Doi:20.4414/smw.2019.20072.
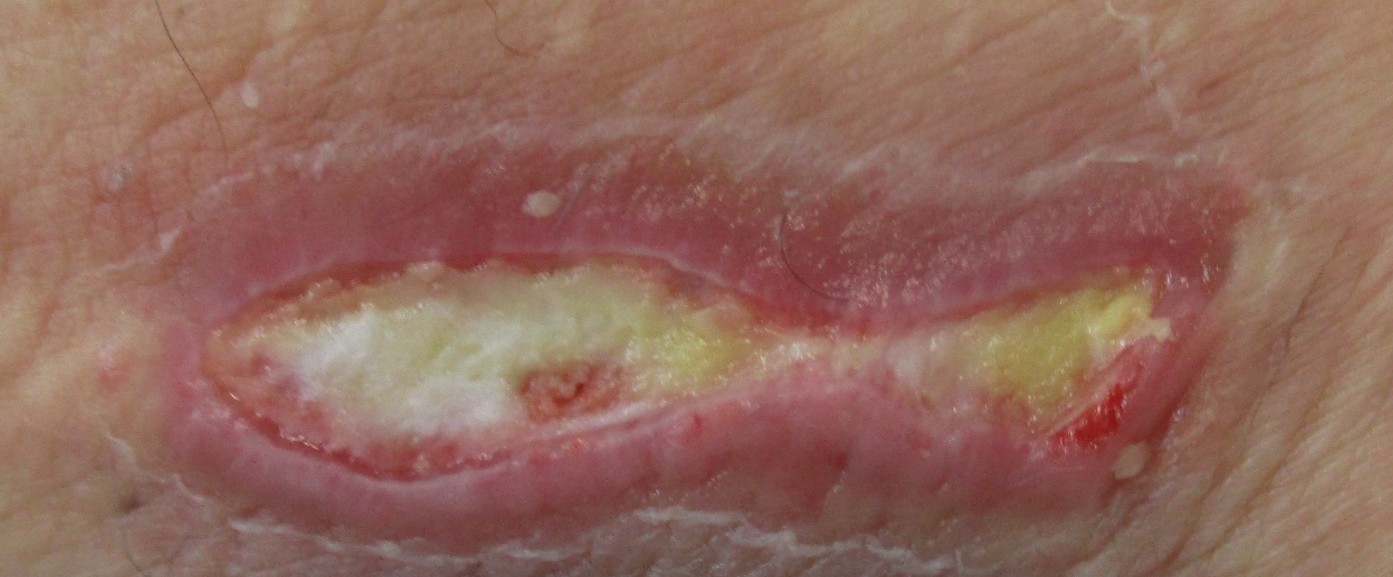
Wochenlange konservative Therapie:
-> Keine Wirkung
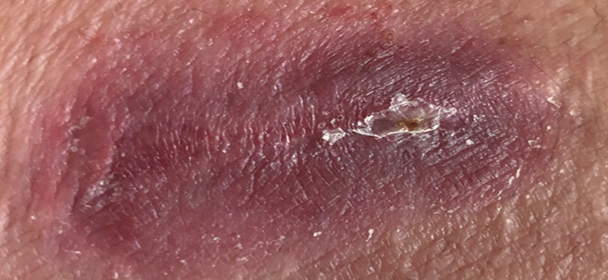
1 Woche nach 1 Spritze Adalimumab:
-> Wunde verschlossen